


The powerful and the damned
That cool ketamine-under-general-anaesthesia trial is underpowered, sorry


Ketamine is an old medication that has been repurposed as a treatment for depression. Proponents argue it is fast-acting, and effective in ‘treatment resistant’ cases. There is evidence from meta-analyses of randomised controlled trials that it is effective in reducing depressive symptoms after a single infusion. The isomer esketamine, given by nasal spray, has been granted FDA approval for treatment resistant depression.
Enthusiasm is not universal though. UK clinical guidelines notably recommended against the use of esketamine. One concern they cite is a lack of blinding in trials due to the dissociative effects of ketamine, coupled with a high expectancy of benefit from participants.
Issues around blinding are not specific to ketamine, it is a problem common to many new medications in psychiatry that have strong psychoactive effects, like psychedelics or MDMA. A true test of a new treatment needs to be randomised (participants randomly allocated to each arm) and blind (neither the participant nor the researcher know the allocation). Blinding is important because, either consciously or subconsciously, if you know you’ve been given the active treatment, you tend to feel better.
This presents a problem for treatments like ketamine - when you take a dose, you know you’ve taken it. Even psychoactive controls, like benzodiazepines, don’t cut the mustard. Most participants correctly guess which arm they’ve been allocated to, which defeats the purpose of a double-blind trial.
Now there is blinding and then there is blinding. A couple of weeks ago, a randomised trial from Boris Heifets’ team took it to the next level (if you can’t access the article, a pre-print version is open access). They utilised the familiarity of ketamine in anaesthetic settings, to give an infusion while patients with depression were unconscious for routine surgery (truly blinded) and found no difference from placebo.
I’ve seen lots of interesting explanations for this negative result, but in this post I’m going to put forward a more prosaic one - the sample size was too small.
Randomised trial of ketamine masked by surgical anaesthesia in patients with depression
Right, let’s get to the compliments; this was a cool study. They used an innovative experimental design and asked an important question. It was an achievement to recruit 40 patients who had moderate depression, who were due to undergo routine surgery, and who were willing to have an infusion of ketamine. The study was well-conducted and analysed appropriately. It was pre-registered. The authors were transparent, providing their code (which was well annotated), anonymous patient data, and their protocol.
The main results look conclusive:
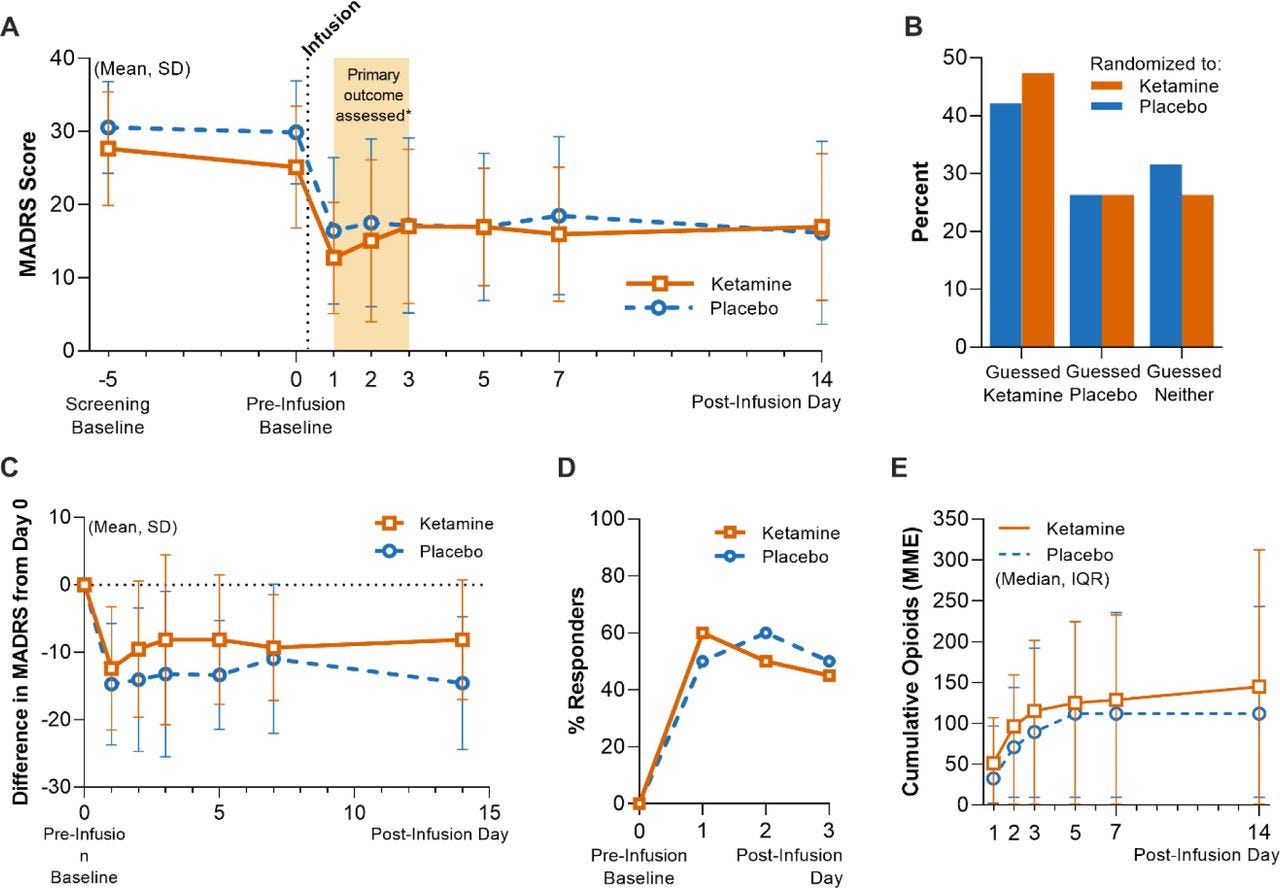
Firstly, as shown in B, blinding was successful - participants were unable to guess their allocation. Less than half of the ketamine group thought they had been given the active treatment, only marginally higher than those in the placebo arm.
Secondly, there were no differences between ketamine and placebo, in terms of depression scores, proportion who responded, or opioid consumption. Essentially no outcomes differed between groups.
Why didn’t ketamine beat placebo? We know from meta-analyses that it is effective for depressive symptoms, so a negative result does require explanation.
From the authors’ discussion and the reaction online there seems to have been three broad takes:
Ketamine requires the expectancy plus acute dissociative effects for antidepressant action. Essentially this means that part/all of the effect is a psychological reaction to the acute administration of ketamine rather than addressing any biological abnormality. When this is inhibited (by the patient being unconscious) there is no antidepressant effect.
General anaesthesia is an antidepressant. Given that both the ketamine and placebo groups had a marked rapid reduction in antidepressant symptoms, could the anaesthetic agents or the process of general anaesthesia be effective in treating depressive symptoms? This seems a little far-fetched to me, without supporting empirical evidence.
Anaesthetic agents block the antidepressant action of ketamine. Again, I don’t think this is rooted in biology, but another proposal is that the antidepressant effect of ketamine was blocked. There is precedent in opioid blockage inhibiting this antidepressant action, though in the trial the dosage of opioids pre-surgery was low.
Of those three, the first seems most likely. However, a simpler explanation has gained less attention - the sample size was too small to detect a meaningful difference between ketamine and placebo.
Power calculations
When presented with a negative trial, we need to consider whether it is a true negative (meaning there really is no difference between treatment and placebo) or a false negative (meaning there is a difference but the trial failed to detect it). The most obvious way to miss a true effect is through the sample size being too small - we call this underpowered.
An antidepressant trial with 20 participants per group is prima facie underpowered. But how can we tell if the sample size was large enough? You can’t just trust a random post you found on the internet! Instead, formal methods are available to calculate the required sample size, known as a power calculation. Part of the protocol and ethical approval for all studies entails a justification of the sample size. Doing a trial that is too small is arguably unethical - it wastes resources and puts patients at risk for no clear benefit.
A power calculation generally requires three parameters: (i) the minimum effect size we wish to detect; (ii) the alpha level (this is the threshold for finding a false positive result and by convention is set to 0.05, meaning that on 5% of occasions we would expect to find a positive result purely by chance; (iii) the power we require to detect a true effect (by convention this is usual set at 80%, meaning if a true effect exists between treatments we would expect to detect it 80% of the time).
Because the authors made their protocol publicly available, we can see their justification for the sample size prior to conducting the study, which I have copied below:
Based on published data from other investigators (Zarate et al., 2006) and our own published work (Williams et al., 2018), we estimate a sample size of 15 patients per group, alpha=0.05, power = 80% to detect a 30% change in depression rating from baseline. We will include an additional 5 patients per group to account for participant dropout, for a total of 20 patients per group.
I have some concerns about this power calculation:
The references are of two small studies Zarate n=17, Williams n=12. These studies are going to have inflated effect sizes - why did they choose these over bigger trials?
The authors don’t state the expected effect size, only that they will detect a 30% change of depressive symptoms from baseline. This doesn’t make sense to me - a power calculation for a placebo controlled trial is explicitly to test the difference between groups, not just the change in symptoms from baseline.
They calculated 15 participants per group (remarkably low). However, they then aim to recruit an additional five participants per group to account for patient dropout. I find it difficult to believe they were concerned that 1/3 of their sample would drop out, given the follow-up for the primary outcome was three days (indeed in the actual study no participants dropped out).
So far, not so good. What did they say about the sample size in the published paper:
Our sample size estimation was derived from an a priori power analysis for the primary outcome. In an RCT of ketamine versus active placebo, participants had a mean decrease of 10.9 points (s.d. 8.9) in MADRS total score relative to pre-infusion scores compared with a mean decrease of 2.8 points (s.d. 3.6) with midazolam15. For reference, the minimum clinically important difference on the MADRS is estimated to range from 3 to 9 points88. Using these results, we computed an estimated total sample size of 38 participants at a two-sided alpha level of 0.05 and 80% power to detect this difference if using parametric testing. An additional two participants were added to account for potential attrition, for a total of 40 participants.
So the power calculation is different in the paper than the protocol. Is this one any better? They’ve used another reference, Phillips et al. 2019 a trial of ketamine versus midazolam (n=41). In terms of 24 hours reduction in symptoms, using the numbers provided above, I calculated an effect size (Standardised Mean Difference) of 1.2. This would be the biggest effect size of any psychological or pharmacological treatment for any psychiatric disorder and is therefore unrealistic.
In the published version, the sample size calculation was 19 patients per group. To get up to that magic number of 40, they proposed recruiting an extra two patients in case of dropouts. Are you starting to feel skeptical?
Let’s work out what an adequate sample size would have been. For this, I’ve taken the effect size from a meta-analysis of intravenous ketamine versus placebo, using the outcome of symptom scores at 24 hours. The SMD is 0.77 (95% Confidence Intervals 0.46-1.08). Using G*Power, the sample size required to detect the lower bound of the Confidence Interval is 152:
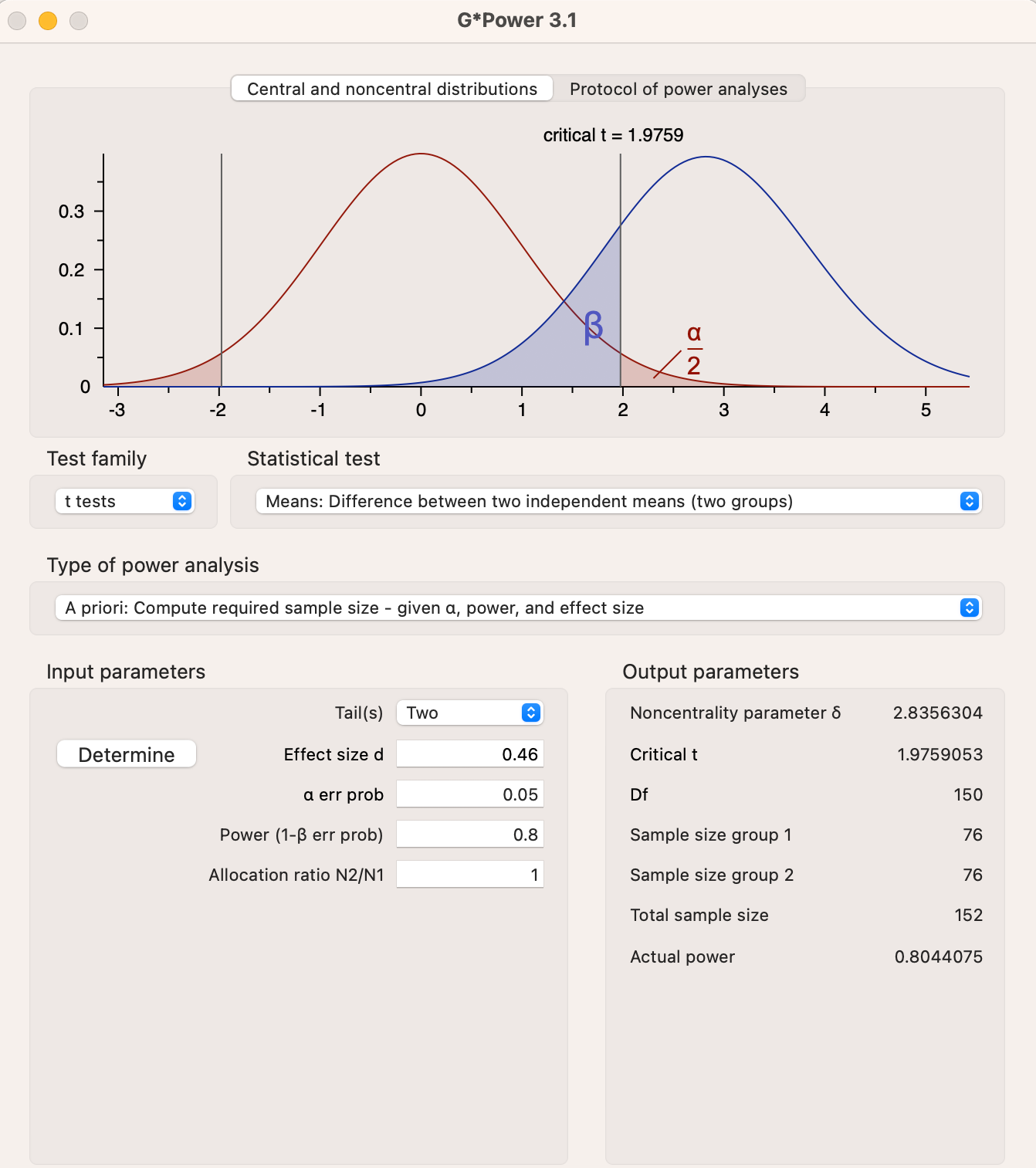
This is roughly four times the actual sample size, indicating the trial was grossly underpowered to detect a realistic effect size - the study was too small to rule out a difference between ketamine and placebo. The lower bound of 0.46 isn’t particularly small for depression, the pooled SMD for all antidepressants is 0.3. Even if we are generous and use the estimate of 0.77, the required sample size is still 56.
A caveat to this discussion is that the researchers didn’t use a simple parametric test like a t test for their analysis, instead using mixed-effect models to test for differences between groups over three days. Although potentially more powerful than simpler tests, these statistical analyses require more complex power calculations. I can’t see any evidence the authors took this into account in their sample size justifications.
Unanswered question
OK, I get it. I’m criticising this from the comfort of my laptop. I get that this trial was hard to recruit, done on institutional funding, not backed by big governmental or pharma funding. I get that it is common practice to fudge power calculations to get a feasible, affordable sample size. I get that power calculations are often pulled out of air as a requirement for ethical approval.
However, unless sample size is carefully justified with a genuine a priori power calculation, we cannot be confident that a negative trial is a true negative. And therefore, whether ketamine is superior to placebo when truly blinded is still up for grabs.
Where does this leave ketamine? I don’t think the study should shift your opinion strongly in either direction. There is replicated evidence that ketamine beats placebo in not-quite-double-blind randomised controlled trials. While in a well-powered (n=400) but completely unblind randomised trial, ketamine was as good as ECT!
Instead, this underpowered study poses a question, does ketamine work with complete blinding? It demonstrates a feasible, innovative methodology (masking with general anaesthesia) to address the question. This opens the door for a future trial to give us a conclusive answer, using an adequately powered sample.
OK, no evidence the authors took into account the mixed-effect model used to test for differences between groups over time in their power calculations, but can't you/someone run the more complex and appropriate power calculations to check how far from reasonable the sample size actually is considering the statistical model used?
One issue worth noting:
ECT is ALSO done under anesthesia.
How do we know that ECT's benefits aren't from anesthesia?
ECT seemed to get a lot better when they started anesthetizing people for the procedure, which is exactly what you'd expect if anesthesia was the active thing here.